Introduction
With the development of high-throughput sequencing technology, emerging single-cell RNA-seq has enabled the measurement of expression of thousands of genes across thousands of individual cells. Therefore, major biological systems have been investigated, transforming our understanding of these fundamental processes.
Challenges and use case
These groundbreaking techniques also came with new big analytical challenges to ensure that the data generated are fully and correctly interpreted. Hence, one of the most crucial problem to circumvent comes from the fact that solid tissues are dissociated into single cells and separated from their native spatial context, such that further analyses lack crucial information on cells’ environments and locations.
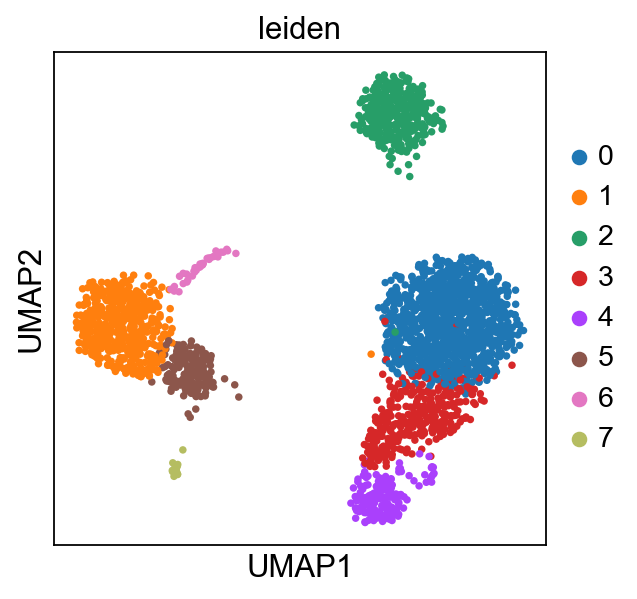
Figure 1. Cluster of cells after dimension reduction using ScanPy. Image ScanPy 2023
To address these questions, a large number of computation approaches have emerged to analyze single-cell RNA-seq data, like Seurat (1), Monocle (2), PAGOLA (3) or Cell Ranger (4). Seurat, the first package developed to handle this type of data, was created by Aviv Regev from the Broad Institute at Harvard. This R package revolutionize the way scientists were working with single-cell RNA-seq data by giving them a way to analyze properly their data. Afterward, multiple packages appeared, the most significant being the Python library ScanPy (5). This article will focus mainly on this library.
History of ScanPy
ScanPy was developed as an open-source python library by the Theis group located at the Helmholtz Munich Research Institute (6). It includes preprocessing, visualization, clustering, pseudotime and trajectory inference, differential expression testing, and simulation of gene regulatory networks. It allows for the identification of cell clusters, that have similar gene expressing pattern, and display these groups of cells in a way that can be easily studied further (Figure 1).
One of the most interesting part of ScanPy is the fact that it is now developed by a full community of scientists, that created a multitude of tools like AnnData (7), to handle data matrices back to ScanPy or ScVI that extends further single-cell analysis into probabilistic models, multimodal analysis, spatial data mapping, Ambient RNA removing and much more (8).
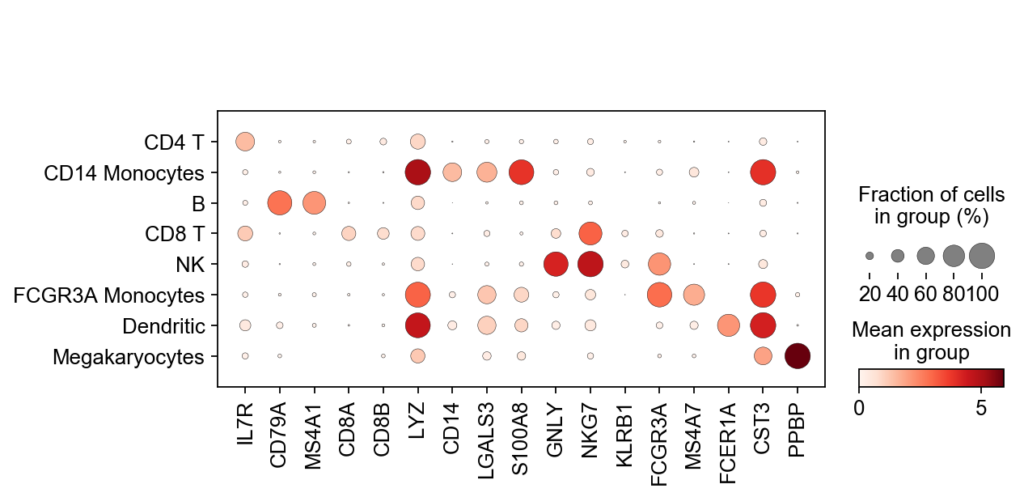
Figure 2. Mean gene expression by cluster. The fraction of cells expressing the gene is also represented. Image ScanPy 2023
After retrieving the data from the sequencer, ScanPy can then applies quality controls and creates clusters of cells (based on dimension reduction using t-SNE or UMAP). These clusters will represent the different type of cells that are represented in the analyzed tissue or disease (Figure 1).
Each clusters could be represented as gene expression values and therefore; by applying known biomarkers, could be identified properly as specific cell types. Hence; after identification; each of these clusters will highlight new underlying mechanisms and subgroup of cells that could have a key role in the tissue or disease being analyzed (Figure 2).
From this type of analysis we could get a complete view of all the genes expressed for each type of cell represented in a particular tissue or disease and therefore determine key genes or gene expression modifications to a specific cell type. This will give the opportunity to specifically act on it or identify a strategy to dampen or treat the effect of a particular disease.
References
- https://satijalab.org/seurat/
- Trapnell, C., Cacchiarelli, D., Grimsby, J. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol32, 381–386 (2014). https://doi.org/10.1038/nbt.2859
- Kharchenko, P., Silberstein, L. & Scadden, D. Bayesian approach to single-cell differential expression analysis. Nat Methods11, 740–742 (2014). https://doi.org/10.1038/nmeth.2967
- Zheng, G., Terry, J., Belgrader, P. et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun8, 14049 (2017). https://doi.org/10.1038/ncomms14049
- https://github.com/scverse/scanpy
- https://scanpy-tutorials.readthedocs.io/en/latest/
- https://anndata.readthedocs.io/en/latest/
- https://scvi-tools.org/



